Портальная гипертензия (ПГ) — синдром, который служит основой тяжелых клинических проявлений цирроза печени (ЦП), таких как формирование варикозно расширенных вен пищевода или желудка и их разрыв, асцит и печеночная энцефалопатия. Основным механизмом повышения давления в воротной вене при ЦП является увеличение внутрипеченочной резистентности к портальному притоку. Морфологическим субстратом ПГ является отложение коллагена в ацинусе печени с сужением синусоидального просвета и, как следствие, уменьшение площади поперечного сечения печеночных синусоид. Помимо этого, увеличение внутрипеченочного портального сопротивления происходит за счет сдавления центролобулярных венул регенераторными узелками, гранулемами и развитием портального воспаления [1].
Помимо структурного компонента, в увеличении печеночной резистентности также участвует и потенциально обратимый, вазоактивный компонент [2]. При циррозе повышается сократительный тонус клеток гладких мышечных волокон и миофибробластов вокруг синусоид и венул печени [3]. Норадреналин, субстанция Р, тромбин, ангиотензин II, эндотелин и простаноиды могут повышать сократительный тонус миофибробластов и, следовательно, портальную резистентность [4, 5]. Однако основным источником динамического повышения внутрипеченочной портальной резистентности является эндотелиальная дисфункция. Снижение биологической доступности оксида азота (NO) в синусоидах и увеличение выработки простаноидов (продуктов метаболизма циклооксигеназы), таких как простагландин H2 и тромбоксан A2, по-видимому, являются основными участниками эндотелиальной дисфункция при ЦП [6, 7].
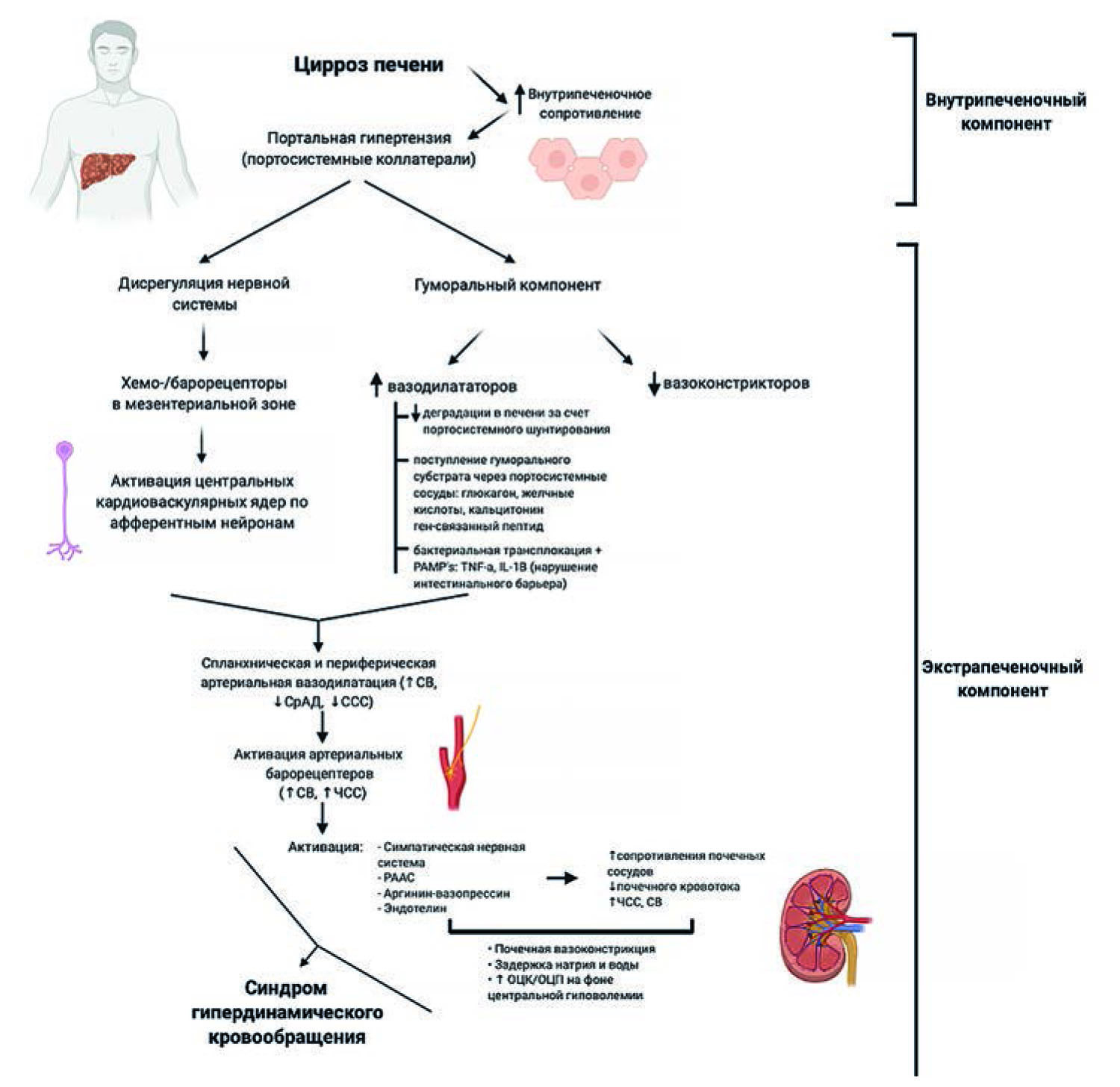
Целью данного обзора является обобщение современных знаний о физиологии спланхнической вазодилатации и синдрома гипердинамического кровообращения (рис. 1), что является необходимым для профилактики и лечения ПГ и ее осложнений. Поиск публикаций для данного обзора проводился с использованием электронных ресурсов электронной библиотеки eLIBRARY.ru, библиографической базы статей по медицинским наукам (MEDlars onLINE) Национальной медицинской библиотеки США (U.S. National Library of Medicine, NLM), базы данных Cochrane Reviews, а также China National Knowledge Infrastructure, WANGFANG DATA с 1966 по 2019 г. Глубина поиска составляла 35 лет. Из обнаруженных 662 источников для обзора литературы были отобраны 67, опубликованные преимущественно за последние 20 лет.
Рис. 1. Патофизиология синдрома гипердинамического кровообращения
Fig. 1. Pathophysiology of hyperdynamic circulation syndrome
Из-за увеличения внутрипеченочной резистентности при ПГ можно было ожидать снижения портального кровотока. Однако несмотря на увеличение диаметра воротной вены и снижение в ней скорости потока крови, портальная гемодинамика при ЦП характеризуется развитием сети коллатералей с увеличением суммарного кровотока в системах воротной, селезеночной и брыжеечных вен [8].
Давление в воротной вене является результатом противодействия между объемной скоростью потока крови, поступающего в портальную систему, и сопротивлением оттоку на уровне цирротически измененной печени. Математическое выражение этой зависимости может быть представлено в виде формулы Ома (1.1):
Р = Q × S (1.1),
Основной детерминантой ПГ при ЦП является увеличение сопротивления портальному потоку, в то время как увеличение притока играет второстепенную роль [9]. В печени с нормальной резистентностью при изменении портального кровотока давление в воротной вене не изменяется [10] из-за высокой степени податливости сосудов портальной системы. При увеличении сопротивления портальному потоку (вследствие ЦП) и снижении податливости сосудов системы воротной вены давление в воротной вене напрямую зависит от величины портального кровотока [11].
Механизмом, объясняющим поддержание высокой скорости объемного кровотока по системе воротной вены, является открытие портосистемных коллатералей, вызванное увеличением сопротивления оттоку от портальной системы. Открытие коллатерального кровообращения происходит посредством реперфузии и дилатации ранее существующих сосудов, а также вследствие генерации новых сосудов, как было показано в экспериментальных исследованиях, демонстрирующих роль ангиогенетических факторов, таких как фактор роста эндотелия сосудов (VEGF — vascular endothelial growth factor), в патогенезе развития коллатералей у крыс с индуцированной ПГ [12]. Портосистемные шунты ответственны за развитие желудочно-кишечного кровотечения, главным образом из-за разрыва варикозно расширенных вен пищевода или желудка. По ним же происходит попадание в системный кровоток ряда метаболитов, которые обычно удаляются печенью. Портосистемные шунты играют основную роль в патогенезе гипердинамического кровообращения, асцита и печеночной энцефалопатии [13].
Увеличение спланхнического кровотока при ПГ вследствие ЦП вызвано вазодилатацией спланхнических артерий селезеночного и мезентериального бассейнов. В последние годы механизмы, ответственные за снижение резистентности мезентериальных артерий были тщательно изучены. В качестве возможных медиаторов были предложены многочисленные вещества и даже целые системы: глюкагон [13, 14], простациклин (PGI2), интестинальный вазоактивный пептид, гистамин, вещество Р, эстрогены, холецистокинин, аммиак, эндотоксины, аденозин, желчные кислоты, NO, пептид, связанный с геном α-кальцитонина, адреномедуллин, VEGF, монооксид углерода, эндогенные каннабиноиды. Эндотелиальные факторы также играют в этом процессе определенную роль [13, 15].
В настоящее время NO рассматривается в качестве основного фактора обусловливающего снижение мезентерального сопротивления при ЦП. NO имеет очень короткий период полувыведения (20–30 с), он свободно диффундирует через клеточную мембрану и действует главным образом за счет стимуляции гуанилатциклазы, выработки циклического гуанозинмонофосфата (цГМФ) с последующим расслаблением клеток гладкой мускулатуры. У пациентов с ПГ вследствие повышения активности эндотелиальной NO-синтетазы (eNOS — Endothelial nitric oxide synthase) биодоступность NO значительно увеличена [16]. Этот факт получил подтверждение в ряде лабораторных экспериментов: у крыс с индуцированной ПГ наблюдалось повышение активности NOS даже в терминальных стадиях заболевания [15]. Данный феномен, по-видимому, обусловлен несколькими механизмами. Провоспалительные цитокины, VEGF и механические силы, такие как напряжение сдвига, индуцируют сигнальные каскады, основной целью которых является активация протеина теплового шока 90 (Hsp90, heat shock protein 90), который, в свою очередь, активирует eNOS [17]. Еще одним механизмом, увеличивающим содержание к крови фактора некроза опухоли, ко-фактора eNOS, и тетрагидробиоптерина является транслокация бактерий в брыжеечные лимфатические узлы из кишечника [18].
При декомпенсированном ЦП в эндотелии брыжеечных артерий активируется еще одна форма NO-синтетазы — индуцированная синтетаза. Впрочем, ее активная роль в увеличении биодоступности NO в брыжеечном сосудистом русле пока не была продемонстрирована [19]. Помимо своего вазодилатирующего действия NO также является фактором ангиогенеза спланхнического циркуляторного русла, что продемонстрировано в ряде экспериментальных наблюдений [20].
Система NO/eNOS не является единственной, участвующей в вазодилатации брыжеечного сосудистого русла при ЦП. Это было доказано в эксперименте у лабораторных крыс с индуцированным ЦП, которым введение ингибиторов NOS лишь частично корректировало спланхническую вазодилатацию [15]. Тем самым было доказано вовлечение других вазоактивных систем в снижение брыжеечной сосудистой резистентности. У пациентов с ЦП также повышается циркуляторный уровень простагландина I2 (PGI2, prostaglandin I2), эндогенного вазодилататора, который, подобно NO, продуцируется клетками сосудистого эндотелия [21]. В экспериментальных условиях у лабораторных крыс с индуцированной ПГ было продемонстрировано усиление экспрессии циклооксигеназы-1. Ингибирование циклооксигеназы-1 индометацином снижало степень ПГ и улучшало гипердинамическое кровообращение [22].
Также в спланхнической дилатации при ЦП участвует конечный продукт катаболизма гема — оксид углерода (СO). CO, подобно NO, является активатором гуанилатциклазы, усиливая выработку цГМФ с последующим расслаблением клеток гладкой мускулатуры [23].
Эндогенные каннабиноиды — распространенные сигнальные молекулы липидной природы, которые реализуют свои центральные и периферические эффекты посредством воздействия на специфические рецепторы CB1 и СB2. Экспериментальные данные свидетельствуют о том, что эндогенные каннабиноиды способствуют развитию спланхнической вазодилатации путем активации CB1-рецепторов в брыжеечной сосудистой сети. Влияние эндогенных каннабиноидов на брыжеечные сосуды, по-видимому, связано как с увеличением продукции NO, так и с неизученным NO-независимым механизмом [24].
Вазодилатация, наблюдаемая у пациентов с ПГ, также является следствием вегетативной дисфункции, вызываемой снижением реактивности к вазоконстрикторным системам, таким как симпатическая нервная система, вазопрессин, ангиотензин II и эндотелин-1.
Вегетативная дисфункция зачастую приписывается полинейропатии, связанной со злоупотреблением алкоголем, однако доказано участие в этом и иных механизмов [15]. Снижение регуляции или уменьшение сродства рецепторов к вазоконстрикторам может объяснить длительную системную и спланхническую вазодилатацию, которая происходит несмотря на компенсаторную активацию вазоконстрикторных систем [25].
При ЦП увеличение кровотока по брыжеечной артерии не является единственным фактором, определяющим степень ПГ. Было отмечено, что у пациентов с ЦП и ПГ селезеночный кровоток увеличивается в большей степени, чем брыжеечный, обеспечивая до 60–70 % портального притока [26]. Такое увеличение селезеночного кровотока при ЦП наблюдается у пациентов со спленомегалией. Помимо этого, существует взаимосвязь между размером селезенки и диаметром воротной вены, а также величиной селезеночного и портального кровотока [27].
Эти данные подтверждают, что спленомегалия и увеличение селезеночного кровотока при ЦП не являются пассивными явлениями, однако они также способствуют поддержанию ПГ путем перегрузки портальной системы объемом циркулирующей по ней крови [26].
Нарушения нормальной цитоархитектоники печени при циррозе приводят к развитию ряда структурных и сосудистых изменений. ПГ, а также артериальная вазодилатация в системном и спланхническом кровообращении при ЦП, часто ассоциированы с синдромом гипердинамической циркуляции (СГЦ). Основными проявлениями СГЦ являются увеличение сердечного выброса, частоты сердечных сокращений (ЧСС) и снижение общего периферического сосудистого сопротивления как ответ на снижение эффективного объема циркулирующей крови (ОЦК). Уменьшение эффективного ОЦК и активация барорецепторов правого предсердия, каротидных синусов и дуги аорты, в свою очередь, активируют симпатическую нервную систему и ренин-ангиотензин-альдостероновую систему, что приводит к задержке натрия и воды с возможным образованием асцита [15].
СГЦ также способствует развитию сердечно-сосудистых нарушений, включая диастолическую дисфункцию, снижение систолической реакции на стресс и ряд электрофизиологических нарушений, которые, проявляясь совместно, получили название цирротической кардиомиопатии [26].
СГЦ при ЦП характеризуется увеличением сердечного выброса и ЧСС, а также снижением общего периферического сопротивления сосудов, что проявляется снижением артериального давления [26]. Главной причиной этого является происходящие при ЦП системная и спланхническая вазодилатация, что в конечном итоге приводит к компенсаторным изменениям в сердечно-сосудистой системе в общем и региональных циркуляторных руслах отдельных органов.
Данный факт доказан в ряде экспериментальных исследований. Введение ингибиторов NOS лабораторным крысам с инсталлированным ЦП приводило к нормализации гемодинамических параметров [15], но только отчасти компенсировало спланхническую вазодилатацию [19]. У нейропептида Y, симпатического ко-трансмиттера норадреналина, обнаружены сходные эффекты в отношении редукции портального кровотока и улучшение гипердинамического состояния посредством спланхнической вазоконстрикции у лабораторных крыс с асцитом [28].
Коллатеральное кровообращение при ЦП способствует развитию СГЦ как напрямую, путем снижения периферического сопротивления потоку крови, так и косвенно, позволяя вазоактивным веществам из кишечника обойти печень и попасть в системный кровоток. Несмотря на снижение системного и висцерального сосудистого сопротивления при ЦП и ПГ, уменьшение резистентности кровотоку наблюдается не во всех региональных сосудистых руслах. Так, снижение кровотока при ПГ происходит в почках [29], головном мозге [30] и мышцах [31]. Чем быстрее прогрессирует печеночная недостаточность при ЦП, тем более выраженно уменьшается приток крови к этим органам [29].
СГЦ является патогенетической основой развития таких осложнений ЦП, как гепаторенальный синдром, гепатопульмональный синдром и тканевая гипоксия. Поскольку для функционирования печени и кишечника требуется около трети всего сердечного выброса, гипердинамическое кровообращение прямо или косвенно способствует развитию и прогрессированию еще двух серьезных осложнений ЦП — асцита и варикозного расширения вен пищевода.
Прямым следствием СГЦ являются и «классические» симптомы печеночной недостаточности: тахикардия, субцианотичная теплая кожа, системная артериальная гипотония.
Следующее важное проявление гипердинамического кровообращения — увеличение ОЦК [32]. Периферическая артериальная вазодилатация вызывает «депонирование» крови в сосудах спланхнической циркуляции, паренхиматозных органах и «емкостных» тканях (мышечная, жировая), вызывая тем самым снижение преднагрузки на сердце. Ответом на снижение преднагрузки является активация ряда нейрогуморальных систем (симпатоадреналовая, ренин-ангиотензин-альдостероновая, вазопрессин и пр.), что приводит к задержке натрия и жидкости, способствуя увеличению ОЦК [15].
Несмотря на увеличение сердечного выброса, у пациентов с ЦП уменьшается амплитуда систолической и диастолической реакции миокарда желудочков на катехоламины и стресс. Обычно это сочетается с гипертрофией или дилатацией желудочков сердца, а также с рядом электрофизиологических нарушений (удлинение интервала QT) [28]. Изначально считалось, что причинной этому является манифестация латентной алкогольной кардиомиопатии, однако более поздние исследования показали схожую картину снижения сократимости сердца и при развитии неалкогольного ЦП, а также в экспериментальных моделях. Таким образом, в настоящее время изменения вентрикулярной сократимости при ЦП получило название «цирротической кардиомиопатии» (ЦКМП). В ее развитии, по-видимому, участвуют такие механизмы, как измененная β-адренэргическая передача сигналов, дисфункция мембраны кардиомиоцитов и избыточное поступление в кровоток при ЦП «прямых» кардиодепрессантов: NO, цитокины и эндоканнабиноиды [33]. Тем не менее, принимая во внимание ассоциацию с системной вазодилатацией, выраженная степень сердечной недостаточности все же не является характерным признаком этого осложнения.
В отсутствии специфических диагностических критериев точная частота развития ЦКМП остается неизвестной. Характерные проявления ЦКМП включают следующие: ослабленную систолическую или диастолическую реакцию на стрессорные стимулы, структурные или гистологические изменения камер сердца, электрофизиологические аномалии и повышение сывороточных маркеров, подтверждающих растяжение камер сердца [28].
Нарушение реактивности сердечно-сосудистой системы при циррозе, вероятно, вызвано комбинацией факторов, включающих физико-химические изменения мембран кардиомиоцитов, ослабление стимулирующих путей и усиление активности ингибирующих систем [33].
Во многих исследованиях уже было показано, что сократимость кардиомиоцитов в основном детерминирована стимуляцией β-адренергической системы. Эта система состоит из рецепторов, гуанин-нуклеотид-связывающего белка (G-протеин) и аденилатциклазы. Катехоламиновая стимуляция β-адренорептора вызывает ряд взаимодействий, приводящих к продукции вторичного мессенджера циклического аденозинмонофосфата. Циклический аденозинмонофосфат является первичным триггером, который вызывает изменения внутриклеточных потоков кальция и приводит к сокращению кардиомиоцита [34]. Функционирование этого механизма у пациентов с циррозом и в лабораторном эксперименте было оценено в различных исследованиях. Chen et al. продемонстрировали снижение плотности β-адренорецепторов у пациентов с ЦП и в лабораторных моделях [35]. Ma et al. показали, что β-адренорепторный сигнальный путь у пациентов с циррозом нарушается на различных уровнях регуляции: начиная от снижения плотности G-протеина на мембранах кардиомиоцитов и заканчивая нарушением активности самого фермента аденилатциклазы [36].
Было обнаружено, что у лабораторных мышей с ЦП ослабляется ответ на стимуляцию мускариновых рецепторов [37]. Стимуляция мускариновых рецепторов вызывает негативный инотропный эффект на сердечную мышцу, уравновешивая тем самым влияние β-адренергической системы. Таким образом, дисбаланс в регуляции β-адренергической системы и мускариновых рецепторов может вносить свой вклад в патогенез негативных инотропных эффектов на миокард.
Жидкотекучесть мембраны — термин, используемый для описания степени свободы перемещений липидных частиц в двойном липидном слое цитоплазматической мембраны [37]. Было продемонстрировано снижение жидкотекучих свойств цитоплазматических мембран кардиомиоцитов и других клеток организма у пациентов с ЦП [36]. Эти изменения оказывают глубокое воздействие на функционирование β-адренорецепторов, которое заключается в нарушении взаимодействия рецептор–лиганд, уменьшении плотности рецепторов и повреждении внутриклеточного пути передачи сигнала. Восстановление нормальных жидкотекучих свойств мембраны кардиомиоцитов в экспериментальных исследованиях приводило к выраженному улучшению функционирования β-адренорецепторов. Нарушение жидкотекучести мембраны также влияет на функционирование мембранных ионных каналов. Исследование продемонстрировало уменьшение функциональной активности двух типов калиевых каналов в кардиомиоцитах желудочков мышей с циррозом, которое потенциально могло бы объяснить пролонгацию интервала QT на электрокардиограмме [38]. Помимо этого, при циррозе может уменьшаться и поступление кальция в клетку, вызывающее нарушение сократимости сердечной мышцы. Кальций поступает в клетку через мембранные кальциевые каналы, накапливаясь в ней и выделяясь во внеклеточное пространство посредством саркоплазматической сети. Дисфункция кальциевых каналов приводит к снижению сократимости кардиомиоцитов, что также было продемонстрировано в ряде лабораторных экспериментов [36, 38].
NO играет важную роль в регуляции системного и коронарного сосудистого тонуса, участвуя, таким образом, в патогенезе различных нарушений сердечно-сосудистой системы, включая ишемическую болезнь сердца [39]. Неселективная блокада NOS N-омега-монометил-L-аргинином усиливает сократительную реакцию кардиомиоцитов желудочков мышей на изопротеренол без каких-либо эффектов на общую сократимость, подтверждая тем самым ингибиторный эффект NO на сократимость сердца. Аналогично в другом исследовании ингибирование синтеза NO посредством N-омега-монометил-L-аргинином восстанавливало сократительную функцию желудочков у мышей с циррозом без выраженного эффекта на животных из контрольной группы [40].
СО является известным вазодилататором, который также оказывает негативное влияние на сократимость сердца посредством взаимодействия с цГМФ [23]. СО стимулирует гуанилатциклазу, вызывая продукцию цГМФ, который, в свою очередь, активизирует фосфориляцию протеинкиназы G и ингибирует поступление кальция в цитозоль кардиомиоцита. Liu et al. исследовали роль СО в патогенезе ЦКМП. Они показали, что транскрипция матричной рибонуклеиновой кислотой и экспрессия протеина HO-1 в желудочках животных с циррозом значительно увеличиваются в сравнении с контрольной группой животных. Также ими продемонстрировано, что подавление гемоксигеназы значительно уменьшает прирост содержания цГМФ и восстанавливает сниженную сократимость изолятов сосочковых мышц лабораторных животных с циррозом без эффекта на изоляты мышц из контрольной группы [41]. Основываясь на этих находках, Liu et al. подтвердили, что активация пути HO/СО также вовлечена в патогенез ЦКМП.
Положительная регуляция каннабиноидного сигнального пути также имеет место при хронических заболеваниях печени [24]. Эндогенные каннабиноиды оказывают негативный инотропный эффект в лабораторных моделях посредством их взаимодействия с ингибирующими рецепторами, связанными с G-протеином CB1 и CB2, приводя к подавлению активности аденилатциклазы и поступления кальция в цитозоль кардиомиоцита. Gaskari et al. показали, что нарушенная сократимость изолятов сосочковой мышцы левого желудочка восстанавливается после преинкубации ее с антагонистом CB1 [41].
Пациенты с ЦКМП в течение длительного времени могут оставаться бессимптомными из-за снижения постнагрузки на сердце, вызванного системной вазодилатацией, что маскирует клинические проявления легкой или умеренной дисфункции сердца. Сердечная недостаточность у пациентов с ЦП зачастую манифестирует клинически после проведения таких вмешательств, как парацентез большого объема асцита, трансъюгулярное внутрипеченочное портосистемное шунтирование (TIPS — Transjugular Intrahepatic Portosystemic Shunt), трансплантация печени или агрессивная трансфузионная терапия для компенсации кровопотери из варикозно расширенных вен пищевода [42].
Систолическая функция левого желудочка при ее оценке в состоянии покоя по фракции выброса левого желудочка обычно является нормальной (55–60 %). Тем не менее во время физической нагрузки, стресс-тесте и прочих состояниях, при которых повышается производительность левого желудочка, у пациентов с ЦП в сравнении с контрольной группой наблюдается менее выраженное увеличение фракции выброса левого желудочка [43]. Подобное нарушение сократительной способности миокарда является результатом снижения функционального резерва миокарда. Также у пациентов с ЦП может наблюдаться «притупление» реакции миокарда на катехоламины в виде менее выраженного в сравнении с контрольной группой увеличения ЧСС [42, 43].
Несмотря на снижение постнагрузки на сердце, приблизительно у 30 % пациентов с ЦП развивается гипертрофия левого желудочка. Эта гипертрофическая реакция миокарда может быть связана с гемодинамической перегрузкой желудочков (механическим стрессом) или активацией нейрогуморальных путей с последующим ремоделированием миокарда и развитием фиброза. Необходимо отметить, что после выполнения ортотопической трансплантации печени (ОТП) происходит быстрая регрессия гипертрофии левого желудочка [44].
Гипертрофия миокарда, развивающаяся при ЦП, уменьшает растяжимость левого желудочка во время диастолы. Следовательно, относительно небольшое увеличение внутрисосудистого объема может приводить к значительному повышению конечно-диастолического давления. Диастолическая дисфункция левого желудочка может быть самым ранним проявлением сердечной недостаточности у пациентов с ЦКМП, что было обнаружено у 56 % пациентов с применением ультразвуковой тканевой допплерографии [42]. Диастолическая дисфункция связана с плохой переносимостью больших сдвигов внутрисосудистой жидкости, происходящих при ОТП [45].
Удлинение интервала QT — широко известный ЭКГ-феномен у пациентов с ЦП и может быть следствием изменения жидкотекучести плазматической мембраны кардиомиоцитов и ухудшения ионного транспорта через калиевые каналы [36, 38]. Считается, что это может быть потенциальной причиной желудочковых аритмий и внезапной сердечной смерти [45]. Выраженность удлинения интервала QT имеет четкую взаимосвязь с тяжестью печеночной недостаточности при ЦП и нормализуется после выполнения ОТП [46]. У пациентов с ЦП также часто имеют место аномальные хронотропные ответы на физиологические и фармакологические стимулы. Несмотря на наличие у многих пациентов с тяжелой печеночной недостаточностью тахикардии, все же большинство их них не способны значительно увеличить ЧСС при определенных физиологических состояниях (например, при сепсисе, кровопотере), что может ухудшить способность сердца поддерживать адекватный метаболическим запросам уровень СВ [42, 43].
Гепаторенальный синдром (ГРС) является уникальной формой острой почечной недостаточности, вызванной выраженной вазоконстрикцией почечных сосудов у пациентов с прогрессирующим ЦП.
ГРС может возникать спонтанно или быть вызван провоцирующим фактором (> 70 % случаев ГРС 1-го типа) [47]. Триггерными событиями для развития ГРС могут быть бактериальная инфекция, высокообъемный парацентез, желудочно-кишечное кровотечение, алкогольный гепатит, а также развитие ЦКМП и уменьшение сердечного выброса [48].
Патофизиология ГРС полностью не определена, однако она включает прогрессирование относительной гиповолемии за счет спланхнической вазодилатации и активацию чрезмерной почечной вазоконстрикции, приводящую к нарушенному почечному кровотоку, задержке натрия и жидкости [47, 49].
Ключевым в постановке диагноза ГРС является исключение остальных возможных причин почечной недостаточности. Быстрая идентификация ГРС уменьшает задержку терапии и ожидаемо позитивно влияет на исход [50].
ГРС 1-го типа — острая и быстро прогрессирующая форма почечной недостаточности, проявляющаяся увеличением сывороточного креатинина > 221 ммоль/л с прогнозируемой летальностью в нескольких недель при отсутствии возможности трансплантации печени [51]. ГРС 2-го типа протекает менее агрессивно, характеризуется увеличением сывороточного креатинина > 133 ммоль/л и обычно связан с рефрактерным асцитом [52].
ГРС связан с значительной морбидностью и летальностью, определяющей негативный прогноз, часто измеряемый неделями [53]. Лучшим выбором в плане долгосрочной выживаемости у таких пациентов является трансплантация печени, а терапией первой линии — вазоконстрикторы, хотя их реальный эффект скорее заключается в увеличении кратковременной выживаемости и возможности быть мостом к трансплантации печени [54]. Совместное назначение аналога вазопрессина, терлипрессина, вместе с альбумином является терапией первой линии при ГРС 1-го и 2-го типов для противодействия спланхнической вазодилатации [51]. Может быть рассмотрено применение других вазоконстрикторов, α-адренергических агонистов, однако на данный момент недостаточно убедительных данных в пользу их применения [55].
TIPS также является возможной терапевтической опцией, улучшающей почечную функцию при ГРС 1-го и 2-го типов, но его применимость ограничена тяжелой печеночной недостаточностью [56]. Фактически TIPS не улучшает значительно выживаемость у пациентов с резко сниженной функцией печени (Child B и C), однако, несмотря на это, некоторые работы показывают положительный эффект TIPS у пациентов с ГРС 2-го типа и 10–12 баллами по Чайлду—Пью, ожидающих трансплантацию печени [51, 57].
Гепатопульмонарный синдром (ГПС) встречается у 5–32 % пациентов с ЦП и определяется наличием заболевания печени и/или ПГ и внутрилегочной вазодилатацией, что ведет к нарушению альвеоло-артериального градиента кислорода (PaO2 < 80 мм рт. ст. и A-aPO2 > 15 мм рт. ст. или > 20 мм рт. ст. для пациентов старше 64 лет) [58].
Патофизиология ГПС на сегодняшний день до конца не определена. Принято считать оксид азота медиатором внутрилегочной вазодилатации у пациентов с ЦП и ГПС, т. к. у них выше уровни выдыхаемого NO в сравнении с пациентами без ГПС, а также в связи с нормализацией уровня выдыхаемого NO после трансплантации печени [59]. У пациентов с ГПС дилатированы капилляры и прекапилляры до 100 µм в диаметре, увеличено число данных дилатированных сосудов и нарушены механизмы их гипоксической вазоконстрикции [60]. Как результат, смешанная венозная кровь быстрее проходит к легочным венам, тем самым увеличивая кровоток в условиях неизмененной вентиляции, что ведет к нарушению вентиляционно-перфузионного соотношения. С прогрессированием ГПС эффективная оксигенация крови в центре дилатированных альвеолярных капилляров становится невозможна при обычной концентрации кислорода, и требуется увеличение фракции вдыхаемого кислорода, что определяет ГПС как физиологический, а не анатомический (справа налево) шунт. Тем не менее попытки фармакологически повлиять на продукцию NO и внутрилегочную вазодилатацию показали противоречивый результат [61]. Также с ГПС ассоциировано наличие полиморфизма единичных нуклеотидов, регулирующих ангиогенез и ремоделирование легочных сосудов [62].
Проявления ГПС у пациентов с ЦП неспецифичны и чаще всего представляют собой диспноэ (до 70 % пациентов) [63]. Заподозрить ГПС при физикальном осмотре позволяет наличие паукообразных ангиом, цианоза и выраженной гипоксемии. Так как изменение положения тела может влиять на распределение легочного кровотока за счет увеличения перфузии дилатированных сосудов в базальных отделах легких, то пациенты могут отмечать ухудшение дыхания в положении сидя/стоя (ортодеоксия и платипноэ) и, напротив, улучшение в положении лежа. Диагностическим критерием ГПС является нарушение оксигенации в условиях внутрилегочной вазодилатации и сопутствующего почечного заболевания и ПГ [64]. Инструментальная диагностика включает контрастную эхокардиографию и легочное перфузионное сканирование с меченными технецием макроагрегатами альбумина для определения внутрилегочной вазодилатации [65]. На данный момент нет эффективной фармакологической терапии ГПС, а единственной терапией выбора является трансплантация печени (полное восстановление газообмена в более чем 80 % случаев) [66]. Пациенты с ЦП и ГПС с PaO2 < 60 мм рт. ст. должны быть ургентно поставлены в лист трансплантации печени независимо от MELD [67].
Увеличение давления в воротной вене при ПГ, являющееся причиной таких осложнений ЦП, как желудочно-кишечные кровотечения, асцит, печеночная энцефалопатия, ГРС и спонтанный бактериальный перитонит, происходит не только за счет повышения внутрипеченочной резистентности к портальному притоку, но и в результате увеличения спланхнического притока крови в портальную сосудистую систему. Усиление спланхнического кровотока обусловлено вазодилатацией спланхнических сосудов, как селезеночного, так и мезентериального кровообращения, и развитием коллатеральной циркуляции. Повышение в спланхническом кровотоке продукции NO рассматривается в качестве основной причины вазодилатации. Тем не менее другие молекулы, такие как PGI2, ФРЭС, CO и эндоканнабиноиды, также играют роль в этом процессе. Спланхническая вазодилатация характеризуется повышением сердечного выброса и ЧСС, а также снижением системного периферического сосудистого сопротивления и артериального давления. Синдром гиперкинетической циркуляции проявляется мультиорганными нарушениями, клиническая манифестация которых при ЦП характеризуется развитием асцита, варикозного расширения вен пищевода, ГРС, ЦКМП, ГПС и др. Понимание патофизиологии вазодилатации и синдрома гипердинамического кровообращения имеет важное значения для профилактики и лечения осложнений ПГ.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Вклад авторов. Катин М.Л., Гурова М.Ю., Прилуцкий П.С., Дзядзько А.М., Руммо О.О. — разработка концепции статьи, получение и анализ фактических данных, написание и редактирование текста статьи, проверка и утверждение текста статьи.